PLANT TISSUE CULTURE
Plant tissue culture, also referred to as cell, in vitro, axenic,
or sterile culture, is an important tool in both basic and applied studies, as
well as in commercial application. Plant tissue culture is the aseptic culture
of cells, tissues, organs and their components under defined physical and
chemical conditions in vitro. The theoretical basis for plant tissue culture
was proposed by Gottlieb Haberlandt
in 1902
Principle:
Totipotency, - The ability to
regenerate the entire organism from a single somatic cell, i.e., trigger the use of the genetic information
present to direct the entire regenerative and developmental programs needed to
create the whole organism from a single cell, Cyto differentiation:
dedifferentiation and re-differentiation are the principles. Dedifferentiation
is the capacity of mature cells to return to meristematic condition and
development of a new growing point
Competency describes the endogenous potential of a given cell or tissue to
develop in a particular way. For example,
as embryogenically competent cells are capable of developing into fully
functional embryos. The opposite is non-competent or morpho-genetically
incapable.
BRIEF HISTORY:
1838 - Schwann and Schleiden put
forward the theory which states that cells are totipotent, and in principle,
are capable of regenerating into a complete plant. Their theory was the foundation
of plant cell and tissue culture
1902 - Haberlandt proposed concept of in vitro cell culture
1904 - Hannig cultured embryos from several cruciferous species
1922 - Kolte and Robbins
successfully cultured root and stem tips respectively
1926 - Went discovered first plant growth hormone –Indole acetic acid
1934 - White introduced vitamin B as growth supplement in tissue culture
media for tomato root tip
1939 - Gautheret, White and Nobecourt established endless
proliferation of callus cultures
1941 - Overbeek was first to add coconut milk for cell division in Datura
1946 - Ball raised whole plants of Lupinus by shoot tip culture
1954 - Muir was first to break callus tissues into single cells
1955 - Skoog and Miller
discovered kinetin as cell division hormone
1957 - Skoog and Miller gave concept of hormonal control (auxin:
cytokinin) of organ formation
1959 - Reinert and Steward
regenerated embryos from callus clumps and cell suspension of carrot (Daucus
carota)
1960 - Cocking was first to isolate protoplast by enzymatic degradation of
cell wall
1960 - Bergmann filtered cell suspension and isolated single cells by
plating
1960 - Kanta and Maheshwari
developed test tube fertilization technique
1962 - Murashige and Skoog developed MS medium with higher salt
concentration
1964 - Guha and Maheshwari
produced first haploid plants from pollen grains of Datura (Anther
culture)
1966 - Steward demonstrated totipotency by regenerating carrot plants from
single cells of tomato
1970 - Power et al.successfully
achieved protoplast fusion
1971 - Takebe et al.regenerated
first plants from protoplasts
1972 - Carlson produced first inter specific hybrid of Nicotiana
tabacum by protoplast fusion
1974 – Reinhard introduced biotransformation in plant tissue cultures-
starting of genetic engineering
1977 - Chilton et al.
successfully integrated Ti plasmid DNA from Agrobacterium tumefaciens in
plants
1978- Melchers et al. carried
out somatic hybridization of tomato and potato resulting in pomato
1981- Larkin and Scowcroft
introduced the term somaclonal variation
1983 - Pelletier et al. conducted intergeneric cytoplasmic hybridization
in Radish and Grape
1984 - Horsh et al. developed transgenic tobacco by transformation with
Agrobacterium
1987 - Klien et al. developed biolistic gene transfer method for plant
transformation
2005
- Rice genome sequenced under International Rice Genome Sequencing Project
ORGANIZATION OF LABORATORY
Any laboratory designed for plant tissue culture or biotechnology
must focus on cleanliness & maintaining of aseptic condition. The essential
7 fundamental matter is the contamination free condition in all steps of the
procedure. Any laboratory, in which tissue culture techniques are performed, regardless
of the specific purpose, must contain a number of basic requirements.
These are:
a) A general washing area
b) A media preparation, sterilization & storage area
c) Environmentally controlled incubators or culture rooms
d) An observation/data collection area
e) Acclimatization area
a.
Washing area:
The washing area should contain good quality basin, large sink
& well drainage facilities. It should have access to dematerialized water
& double distilled water. Space for drying ovens or racks, automated
dishwashers, acid baths, pipette washers & driers & storage cabinets
should also be available in the washing area.
General guidelines for washing area;
1.
Reusable glassware for tissue
culture should be emptied immediately & need to be soaked in water. Media
or agar must never be allowed to dry on the glassware
2. All glassware containing corrosive chemicals or fixatives should
be separated from the rest of the tissue culture glassware.
3. All glasswares contaminated or coming into contact with microorganisms
should be autoclaved before washing.
4. The contents of any containers should be discarded immediately
after completion of an experiment.
5. Flaks or beakers used for agar based media should be rinsed
immediately after dispensing the media into culture vassels so as to prevent
drying of the residual agar in the beaker prior to washing.
b. Media Preparation Area:
This area comprises the central section of the laboratory, home to most of the activities. This area should have ample storage space for the chemicals, culture vessels & glassware required for media preparation & dispensing. The general laboratory section includes the area for media preparation for autoclaving the media & also for many of the activities that relate to the handling of tissue culture materials. Laboratory equipments required for media preparation room are as followsGas, water & electric supplies & compressed air & vacuum line.
- Water
heater
- Different
types of glasswares
- Hot
plate with magnetic stirrer
- Coarse
& sensitive balance
- Spatula
for use during weighing
- Microwave
oven for rapid heating media & agar mixture
- pH
meter
- Distillation
unit
- De-ionizer
- Metal
racks for holding test tubes in the autoclave
- Test
tubes, flasks, plastic containers
- Autoclave
or cooker
- Storage
tank for distilled and /or de-ionized water.
CHEMICALS FOR CULTURE MEDIA:
A. Inorganic elements:
a. Macro nutrient:
The need of macro nutrients is higher in tissue culture media. It
provides both anion & cation for the plant cell. The name of each element
with available form & important functions are given below:
|
Sl. No. |
Name of the macro nutrient |
Available form |
Function |
|
1. |
Nitrogen (N) |
KNO3 ,NH4 NO3 |
Both structural &
functional role in protein synthesis |
|
2. |
Phosphorus (P) |
KH2PO4 |
Activation in nucleotide
synthesis |
|
3. |
Potassium (K) |
KNO3 |
Essential for activation of
many enzymes, maintenance of ionic balance
of the cell. |
|
4. |
Calcium (Ca) |
CaCl2.2H2O |
Acts as a cofactor &
largely bound to the cell wall & cell membrane,
Essential for cation- anion balance by
counteracting organic inorganic anions. |
|
5. |
Magnessium (Mg) |
MgSO4.7H2O |
Essential for photosynthesis
& many other enzymatic reactions. |
|
6. |
Sulphur (S) |
MgSO4.7H2O,K2SO4 |
Functional role in protein
synthesis. |
b. Micro nutrients
Micro nutrient is essential for plant cell tissue growth. The name
of elements, available salt combination & function are given below:
|
Sl. No. |
Name |
Available form |
Function |
|
1. |
Zinc (Zn) |
ZnSO4.7H2O |
Act as a component of a
number of enzymes, plays active role in protein
synthesis, specially in the synthesis of tryptophan |
|
2. |
Manganese (Mn) |
MnSO4.4H2O |
Help in photosynthesis |
|
3. |
Copper (Cu) |
CuSO4.5H2O |
Plays an important role in
electron transport chain at the time of photosynthesis |
|
4. |
Molybdenum (Mo) |
|
It participates in the
conversion of nitrate to ammonium |
|
5. |
Boron (B) |
H3BO3 |
It is required for the
synthesis of cell wall & cell membrane |
|
6. |
Iron (Fe) |
FeSO4.5H2O |
Formation of protein,
important for biosynthesis of chlorophyll |
|
7. |
Cobalt (Co) |
CoCl2. 6H2O |
Helpful for nitrogen
fixation |
|
8. |
Chlorine (Cl) |
CaCl2. 2H2O |
To control the
osmoregulation of cell development. |
B. Organic Components
a. Vitamins:
Normally plants synthesis vitamins endogenously. When plant cells
& tissues are grown on in vitro condition some essential vitamins are
absolutely required.
|
Sl. No. |
Name of the vitamin |
|
1. |
Thiamine (vitamin-B1) |
|
2. |
Nicotinic acid B20 |
|
3. |
Pyridoxin-HCl (B6) |
|
4. |
Folic acid |
|
5. |
Biotin -Promotion of cell growth & development. |
|
6. |
Riboflavin |
|
7. |
Retinol (vitamin-A) - |
b.Myo-inositol:
It has several functions like sugar transport, carbohydrate
metabolism, membrane structure & cell wall formation.
c. Sugar:
It can be supplied in the form of sucrose, glucose, and fructose.
It is a source of carbon.
d. Amino acid:
Cultured tissues are normally capable of synthesis of amino acid.
In spite of this, the addition of amino acids to the media is important for
stimulating cell growth. Unlike inorganic nitrogen, amino acids are taken up
more rapidly by plant cells. Glycine is the most common amino
acid used in different tissue culture media. Some of the other amino acids
like glutamine, asparagines, cystine etc. are also required for cell culture.
e. Plant growth regulators:
Plant growth regulators are the organic molecules which have
different regulatory effects on growth & development in whole plants &
plant tissues. It is the most critical component of any culture media accepted
that without regulators, in vitro culture is often impossible. Plant growth regulators
which are often used in plant tissue culture are the following.
i. Auxin: The major functions of auxin are cell division, cell
elongation, organogenesis. It is frequently used as a rooting hormone. The
most frequently employed auxins are IAA (Indole-3- acetic acid), IBA
(Indole-3-butyric acid), NAA (Napthalene acetic acid), 2, 4-D (2 ,4- Dichlorophenoxy
acetic acid). IAA is a naturally occurring auxin is added in concentration of 0.01-10
mg/l. The most effective auxin of callus proliferation for most cultures is 2,
4-D, but unfortunately it strongly suppresses organogenesis & should not
be used in experiments involving root & shoot initiation.
ii. Cytokinin: Cytokinins are derivatives of adenine, which promote cell
division, regulate growth and development in plant tissues. It is known as
shooting hormone essential for induction of auxillary branching and
adventitious shoot formation. The most widely used cytokinins are kinetin,
zeatin, BAP (Benzyladenine), 2iP (2- isopentenyladenine).
iii. Other regulators: Other types of hormones which may be used in plant tissue
culture include gibberellins (GA3), which promotes shoot elongation, and
internodal elongation, ethylene and abscisic acid.
Aseptic Transfer Area/Inoculation room:
All the activities of sterile transfers are performed in this
room. There must be a laminar air flow cabinet where all the precautions
should be taken to prevent entry of any contaminant into the culture vial
during the process of inoculation or subculture. Laminar air flow hoods are usually
sterilized by switching on the hood and wipping the working sueface with 70%
ethyl alcohol for 15 minutes before initiating any operation under the hood.
Ultraviolet light (UV) is sometimes installed to disinfect the area; this
light should only be used when people and plant materials are not in the room.
This room is provided with:
1. Laminar air flow cabinet: Inoculation &
subculture by maintaining aseptic condition.
2.
Steribed sterilizer, Sprit lamp/Bunsen burner: Sterilization of the
knives, scalpels, forceps etc.
3.
Stereo-microscope: observe for specific part.
4.
Ethyl alcohol: sterilization and flaming of small instruments.
5. Tiles/glass plates use
during sterile cutting.
6.
Hypochloride solution: sterilization of plant material.
c. Incubation Room/Culture Room: This is the room where light, temperature, humidity is maintained.
All of these environmental considerations will vary depending on the size of
the growth room.
Temperature: is an important
consideration for the tissue culture and other factors like light, relative
humidity, and shelving depend on it. Generally, temp. of the growth room remains
in the range of 25± 2oC. Temp. in the primary growth room can be
maintained by air conditioner.
Lighting facility: Intensity
of light in the room can easily be maintained by using fluorescent light with
timer. However, most culture rooms are lighted at the 1000 lux (for 1000cft)
with some going up 5000-10000 lux.
Light duration: 16-18 h/day.
Light quality: Spectral quality of
light received by in vitro cultures is very important.
Relative humidity: Relative
humidity (RH) is very difficult to control inside the room but humidifier can
be used to control humidity. Humidity inside the room should be 70-75%
Shelves: Shelving with primary
growth rooms can vary depending upon the situations & explants grown. Wood
is recommended for the inexpensive easy to build shelves.
This room is provided with
1.
Temperature control (25± 2oC)
2.
Electricity supply essential
for lighting, cooling and heating
3.
Shelves for culture racks
4.
Fluorescent tubes for
lighting
5.
Timer for regulating day
length
6.
Racks for culture vials
7.
Rotary shaker for suspension
cultures
8.
Observations table.
d. Data collection Area: Culture
room is prepared by glass wall. Qualitative data could be collected from outside
of the culture room through the glass wall. The quantitative data could be collected
from inside the culture room by following aseptic rules and regulation.
e. Acclimatization area:
Plants regenerated from in vitro tissue cultures are transplanted
to vermiculite pots. The potted plants are ultimately transferred to
greenhouses or growth cabinets and maintained for further observations under
controlled conditions of light, temperature and humidity.
Major equipment and their function
|
Sl. No. |
Name of the equipment |
Function |
|
1. |
Autoclave machine, Pressure cooker |
Sterilization of media,
glassware &small instrument. |
|
2. |
Balance |
Measurement of chemical
from the range of µgm to Kg |
|
3. |
Hot plate magnetic stirrer |
To mix the chemical &
other ingredient of media |
|
4. |
pH
meter |
To
determine the pH of various chemicals & media |
|
5. |
Refrigerator |
To
store all sorts of temperature-sensitive chemical & stock
solution. |
|
6. |
Micro
oven |
To
melt agar, agarose & other gelling agents. |
|
7. |
Hot
air oven |
For
dry heat sterilization of cell & suspension culture |
|
8. |
Shaker |
Use
for gentle rotation of cell& suspension culture |
|
9. |
Filter
sterilization unit with vacuum
pump |
Filtration
of thermoliable compound like growth regulator,
vitamin, amino acid etc. |
|
10. |
Microscope |
To
study the cell & tissue culture material at different stages
of development |
|
11. |
Luxmeter |
To
measure the light intensity of the culture room |
|
12. |
Thermometer |
To
record the temperature reading of laboratory & culture
room |
|
13. |
Centrifuge
machine |
To
sediment cell & clean supernatant |
|
14. |
Laminar
air flow cabinet |
To
avoid air remaining contaminant |
Safety rules:1. A laboratory should have an inventory & a complete up-to-date record of all the equipment along with their operating manual.
2. A laboratory should have an inventory & a complete up-to-date record of all the chemicals including the name of manufacturer & grade.
3. All chemicals should be assigned to specific areas preferably by their alphabetical order.
4. Strong acid & bases should be stored separately.
5. Special handling or storage procedure should be posted in the records so that retrieving of chemical is easy, because chemicals need storage at different temperatures ( for example room temperature 4o, -20o C)
6. Chloroform, alcohol, phenol, which is volatile or toxic in nature, must be stored in a fume hood.
7. Chemicals which are hygroscopic in nature must be stored in desiccators in order to avoid caking.
8. Chemicals kept in refrigerator or freezers should be arranged either alphabetically or in small baskets.
1. Eating, smoking and drinking is strictly prohibited in the tissue culture laboratory.
2. Toxic chemical must be handled with appropriate precautions and should be discarded into separate labeled containers. e.g. Organic compounds, halogens etc.
3. Broken glass and scalpel blades must be disposed into individual marked containers.
4. Pipettes, tips, Pasteur pipettes and other things used in the lab should be first collected in autoclavable bags and then it should be finally autoclaved and disposed in safe place.
5. Pipetting any solution should not be conducted without using any pipette.
6. First aid kits should be placed in every laboratory and every individual working in the laboratory should know its location and how to use its contents.
7. Fire extinguishers should be provided in each laboratory.
Steps involved in general techniques.
Regeneration of Plantlets:
1. Preparation of Suitable Nutrient Medium:
Suitable nutrient medium as per objective of
culture is prepared and transferred into suitable
containers.
2. Selection of Explants:
Selection of explants such as shoot tip should be done.
3. Sterilization/ surface decontamination of Explants:
Surface sterilization of the explants by
disinfectants and then washing the explants with sterile distilled water is essential.
4. Inoculation:
Inoculation (transfer) of the explants into the
suitable nutrient medium (which is sterilized by filter-sterilized to avoid
microbial contamination) in culture vessels under sterile conditions is done.
5. Incubation:
Growing the culture in the growth chamber or
plant tissue culture room, having the appropriate physical condition (i.e.,
artificial light; 16 hours of photoperiod), temperature (-26°C) and relative humidity (50-60%) is required.
6. Regeneration:
Regeneration of plants from cultured plant tissues is carried
out.
7. Hardening:
Hardening is gradual exposure of plantlets to an environmental
condition.
8. Plantlet Transfer:
After hardening plantlets transferred to the
green house or field conditions following acclimatization
(hardening) of regenerated plants.
METODS OF STERILIZATION
The various methods of sterilization are:
1. Physical Method
(a) Thermal (Heat) methods
(b) Radiation method
(c) Filtration method
2. Chemical Method
3. Gaseous method


Nutrient medium
Culture media are largely responsible for the in vitro growth and
morphogenesis of plant tissues. The success of
the plant tissue culture depends on the choice of the nutrient medium. In
fact, the cells of most plant cells can be
grown in culture media. Basically, the plant tissue culture media should contain the same nutrients as
required by the whole plant. It may be noted that plants in nature can synthesize their own food material. However,
plants growing in vitro are mainly heterotrophic i.e. they cannot
synthesize their own food.
Composition of Media:
The composition of the culture media is primarily dependent on
two parameters:
1. The particular species of the plant.
2. The type of material used for culture i.e. cells, tissues,
organs, protoplasts.
Thus, the composition of a medium is formulated considering the
specific requirements of a given culture
system. The media used may be solid (solid medium) or liquid (liquid medium)
in nature. The selection of solid or
liquid medium is dependent on the better response of a culture.
Major Types of Media:
The composition of the most commonly used tissue culture media is
given in the following Table, and briefly described below.
White’s medium:
This is one of the earliest plant tissue culture media developed
for root culture.
MS medium:
Murashige and Skoog (MS)
originally formulated a medium to induce organogenesis, and regeneration of plants in cultured tissues. These
days, MS medium is widely used for many types of culture systems.
B5 medium:
Developed by Gamborg, B5 medium was originally designed for cell suspension and callus cultures.
At present with certain modifications, this medium is used for protoplast
culture.
N6 medium:
Chu formulated this medium and it is used for cereal anther culture,
besides other tissue cultures.
Nitsch’s medium:
This medium was developed by Nitsch and Nitsch and frequently
used for anther cultures. Among the media referred above, MS medium is most
frequently used in plant tissue culture work due to its success with several
plant species and culture systems.

Synthetic and natural media:
When a medium is composed of chemically defined components, it is
referred to as a synthetic medium. On the other hand, if a medium contains chemically
undefined compounds (e.g., vegetable extract, fruit juice, plant extract), it
is regarded as a natural medium. Synthetic media have almost replaced the
natural media for tissue culture.
Expression of concentrations in media:
The concentrations of inorganic and organic constituents in
culture media are usually expressed as mass
values (mg/l or ppm or mg I-1). However, as per the recommendations of the
International Association of Plant Physiology,
the concentrations of macronutrients should be expressed as mmol/l–and
micronutrients as µmol/l–.
Constituents of Media:
Many elements are needed
for plant nutrition and their physiological functions. Thus, these elements
have to be supplied in the culture medium to support adequate growth of cultures
in vitro. A selected list of the elements and their functions in
plants is given in the Table below.
Selected list of elements and their functions in plants

The culture media usually contain the following constituents:
1. Inorganic nutrients
2. Carbon and energy sources
3. Organic supplements
4. Growth regulators
5. Solidifying agents
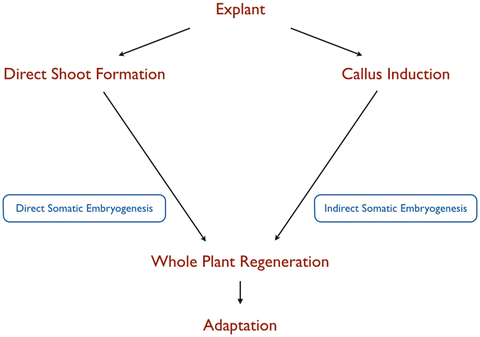
Culture initiation and regeneration through different pathways.
Types of in vitro culture:
1. culture of intact plants e.g. seed culture in orchids
2. embryo culture e.g. immature embryo culture
3. organ culture e.g. meristem culture, shoot tip culture root culture anther culture
4. callus culture
5. single cell culture
6. Protoplast culture.
Culture initiation: selection
of explants, sterilization, media optimization and establishment of the plants
from in vivo to in vitro
Organogenesis
This is a major path of regeneration that
involves the differentiation of culture cells or callus tissue into organs
such as shoot and roots. Plant regeneration through the formation of shoots
and roots is known as plant regeneration through organogenesis. Organogenesis
can occur directly or indirectly from the explants depending on the hormonal
combination of the medium and the physiological state of the explants. Miller
and Skoog demonstrated that the initial formation of roots or shoots on the
cultured callus or explant tissue depends on the relative concentration of auxins
and cytokinins in the culture media. Medium supplemented with relatively high
auxin concentration will promote root formation on the explants and high
cytokinin concentration will promote shoot differentiation. In tissue culture
practices there may be three types of medium in relative combinations of
auxins and cytokinins, which promote either the shoot formation or root formation
or both simultaneously. In the latter case, we can get the complete plantlets,
having both shoot and roots, which can be directly transferred to the pots in
the greenhouse. Whereas in other cases, after the formation of shoots,
individual shoots are transferred to the rooting medium, which promote root
formation. The rooted plantlets can be transferred to a greenhouse for
acclimatization. Plant regeneration through organogenesis is commonly used for
mass multiplication, for micropropagation, and for conservation of germplasm
at either normal or subzero temperatures (cryopreservation)
Skoog and Miller (1957) were responsible
to recognize the regulatory mechanism as a balance between auxin and cytokinin.
As per their finding, a relatively high level of auxin to cytokinin favoured
root formation and the reverse favoured shoot formation. Using this concept,
it has now become possible to achieve organogenesis in a large number of plant
species by culturing explants, calli and cell suspension in a defined medium.
In organogenesis, the shoot or root may form first depending upon the nature
of growth hormones in the basal medium. The genesis of shoot and root from the
explants or calli is termed as caulogenesis
(caulm = stem) and rhizogenesis
(rhizo = root) respectively.
Organogenesis leading to complete plantlet regeneration is a
multistage process consisting of at least three distinct stages.
1. shoot bud formation, 2. shoot development and multiplication 3. rooting of developed shoots.
Caulogenesis is a type of organogenesis by which only
adventitious shoot bud initiation takes place in the callus tissue. When
organogenesis leads to root development, then it is known as rhizogenesis. Abnormal structures
developed during organogenesis are called organoids.
The localized meristematic cells on a callus which give rise to shoots and/or
roots are termed as meristemoids.
Meristemoids are characterized as an aggregation of meristem-like cells. These
can occur directly on an explant or indirectly via callus.
Thus, there are two kinds of organogenesis. A developmental
sequence involving an intervening callus stage is termed 'indirect' organogenesis: Primary explant → callus
→ meristemoid
→ organ
primordium. Direct
organogenesis is accomplished without an intervening proliferate
callus stage: Primary explant → meristemoid → organ primordium. In vitro plant tissues may produce many types
of primordia (adventitious buds and organs) including those that will
eventually differentiate into embryos, flowers, leaves, shoots, and roots.
These primordia originate de novo from a cellular dedifferentiation process,
followed by initiation of a series of events that results in to an organ.
Embryogenesis/ Somatic embryogenesis
This is another major path of
regeneration and development of plantlets for micropropagation or mass
multiplication of specific plants. The cells, under a particular hormonal
combination, change into the physiological state similar to zygotes (somatic
zygotes) and follow an embryonic path of development to form somatic embryos.
These somatic embryos are similar to normal embryos (seed embryos) developed
from zygotes formed by sexual fertilization. The somatic embryos can develop
into a complete plant. Since somatic embryos can germinate into a complete
plant, these can be used for the production of artificial seeds. Somatic
embryos developed by tissue or cell cultures can be entrapped in certain inert
polymers such as calcium alginate and
used as artificial seeds. Since the production of artificial seed is amenable
to mechanization and for bioreactors, it can be produced in large numbers.
Embryogenesis Embryos have been
classified into two categories: zygotic
embryos and non-zygotic embryos.
Zygotic embryogenesis Embryos developing from zygotes (resulting from regular
fusion of egg) are called as zygotic embryos or often simply embryos.
Non-zygotic embryogenesis Usually non- zygotic embryos are formed by cells
other than the zygote. E.g. Parthenogenetic
embryos - formed from unfertilized eggs or a fertilized egg without
karyogamy. Androgenetic embryos – formed
from microspores, micro-gametophytes or sperm.
Somatic embryos (also
called as embryoids, accessory embryos, adventitious embryos and supernumerary
embryos) – formed by somatic cells either in vivo or in vitro. A somatic embryo is an embryo derived from
a somatic cell, other than zygote, usually on in vitro culture. The
process of somatic embryo development is called as somatic embryogenesis.
Stages in development of somatic embryos
Somatic embryos generally originate from
single cells which divide to form a group of meristematic cells. Usually, this
multi-cellular group becomes isolated by breaking cytoplasmic connections with
the other cells around it and subsequently by cutinization of the outer walls
of this differentiating cell mass. The cells of meristematic mass continue to divide
to give rise to globular (round ball
shaped), heart-shaped, torpedo and cotyledonary stages. Somatic
embryo genesis begins with active division of cells which leads to increase in
size but retains the spherical shape. At this stage the primary meristem
(protoderm, ground meristem and procambium) becomes visible. Following this
stage, the callus continues to divide and differentiate into a heart-shaped
embryo, with initiation of cotyledon primordia.
As the cotyledon develops the embryo
passes into the torpedo-shaped stage. The cells inside the cotyledonary ring
divide to form shoot and root apical meristem and procambium differentiation
takes place. In general, the essential features of somatic embryo development,
especially after the globular stage, are comparable to those of zygotic
embryo. The somatic embryogenesis can also be either direct or indirect
depending up on the hormonal composition.

Diagrammatic representation on direct and indirect regeneration
Micropropagation
Micropropagation
is the practice of rapidly multiplying stock plant material to produce a large number of progeny plants, using modern
plant tissue culture methods
Technique of Micro
propagation:
Micro
propagation is a complicated process and mainly involves 3 stages (I, II and
III). Some authors add two
more stages (stage 0 and IV) for more comprehensive representation of micro- propagation. All these stages are
represented in the following Figure, and briefly described hereunder.

Major
stages involved in micropropagation
This
is the initial step in micro- propagation, and involves the selection and
growth of stock plants for about
3 months under controlled conditions.
In this stage, the initiation and establishment of culture in a suitable medium is achieved. Selection of appropriate explants is important. The most commonly used explants are organs, shoot tips and axillary buds. The chosen explant is surface sterilized and washed before use.
It is
in this stage; the major activity of micro propagation occurs in a defined
culture medium. Stage II
mainly involves multiplication of shoots or rapid embryo formation from the explant.
This
stage involves the transfer of shoots to a medium for rapid development into shoots. Sometimes, the shoots are directly
planted in soil to develop roots. In vitro rooting of shoots is preferred while simultaneously handling a
large number of species.
This
stage involves the establishment of plantlets in soil. This is done by
transferring the plantlets of stage III
from the laboratory to the environment of greenhouse. For some plant species, stage III is skipped, and
un-rooted stage II shoots are planted in pots or in suitable compost mixture.
1. Multiplication
by axillary buds/apical shoots.
2. Multiplication
by adventitious shoots.
3. Organogenesis: The
formation of individual organs such as shoots, roots, directly from an explant (lacking preformed meristem) or
from the callus and cell culture induced from the explant.
4. Somatic embryogenesis: The
regeneration of embryos from somatic cells, tissues or organs.
Apical
meristem is a dome of tissue located at the extreme tip of a shoot. The apical meristem along with the young leaf primordia
constitutes the shoot apex. For the development of disease-free plants, meristem tips should
be cultured.
The most widely used media for meristem culture are MS medium and White’s medium. A diagrammatic representation of shoot tip (or meristem) culture in micro propagation is given in Fig and briefly described hereunder.

Diagrammatic representation of shoot tip (or meristem) culture in micropropagation; I, II, III are stages.
In stage I, the culture of meristem is established. Addition of growth regulators namely cytokinins (kinetin, BA) and auxins (NAA or IBA) will support the growth and development.
In stage II,
shoot development along with axillary shoot proliferation occurs. High levels
of cytokinins are
required for this purpose.
Stage
III is associated with rooting of shoots and further growth of
plantlet. The root formation is facilitated
by low cytokinin and high auxin concentration. This is opposite to shoot
formation since high level of cytokinins is required (in stage II).
Consequently, stage II medium and stage
III medium should be different in composition. The optimal
temperature for culture is in the
range of 20-28°C (for majority 24-26°C). Lower light intensity is
more appropriate for good micro
propagation.
The
plant buds possess quiescent or active meristems depending on the
physiological state of the plant. Two types of bud cultures are
used— single node culture and axillary bud
culture.
This
is a natural method for vegetative propagation of plants both in vivo and in
vitro conditions. The bud
found in the axil of leaf is comparable to the stem tip, for its ability in
micro propagation. A bud
along with a piece of stem is isolated and cultured to develop into a plantlet. Closed buds are used to reduce the chances of infections.
A diagrammatic representation of single node culture is below. In single node culture, no cytokinin is added.

Axillary bud culture:
In
this method, a shoot tip along with axillary bud is isolated. The cultures are
carried out with high cytokinin
concentration. As a result of this, apical dominance stops and axillary buds develop. A schematic representation of axillary bud culture for a
rosette plant and an elongate plant
is given below

Fig. Schematic Representation of Axillary Bud Method of Vegetatively Propagating Plants. (a) Rosettle plants; (b) Elongate plants showing bud culture and single node culture.
For a good axillary bud culture, the cytokinin/ auxin ratio is around 10: 1. This is however, variable and depends on the nature of the plant species and the developmental stage of the explant used. In general, juvenile explants require less cytokinin compared to adult explants. Sometimes, the presence of apical meristem may interfere with axillary shoot development. In such a case, it has to be removed.
Organogenesis
is the process of morphogenesis involving the formation of plant organs i.e. shoots, roots, flowers, buds from explant or cultured plant
tissues. It is of two types — direct
organogenesis and indirect organogenesis.
Tissues
from leaves, stems, roots and inflorescences can be directly cultured to
produce plant organs. In direct
organogenesis, the tissue undergoes morphogenesis without going through a callus or suspension cell culture stage. The term direct
adventitious organ formation is also
used for direct organogenesis.
Indirect Organogenesis:
When
the organogenesis occurs through callus or suspension cell culture formation,
it is regarded as indirect
organogenesis. Callus growth can be established from many explants (leaves, roots, cotyledons, stems, flower petals etc.) for
subsequent organogenesis.

Fig: Plant Regeneration Pathways
Micropropagation
of plants by direct and indirect organogenesis
For indirect organogenesis, the cultures may be grown in liquid medium or solid medium. Many culture media (MS, B5 White’s etc.) can be used in organogenesis. The concentration of growth regulators in the medium is critical for organogenesis.
By varying the concentrations of auxins and cytokinins, in vitro organogenesis can be manipulated:
i.
Low auxin and low cytokinin concentration will induce callus
formation.
ii.
Low auxin and high cytokinin concentration will promote shoot organogenesis
from callus.
iii.
High auxin and low cytokinin concentration will induce root
formation.
4.
Somatic Embryogenesis:
The
process of regeneration of embryos from somatic cells, tissues or organs is
regarded as somatic (or asexual)
embryogenesis. Somatic embryogenesis may result in non-zygotic embryos or somatic embryos (directly formed from somatic organs),
parthogenetic embryos (formed from
unfertilized egg) and androgenic embryos (formed from male gametophyte).
Development
of somatic embryos can be done in plant cultures using somatic cells, particularly epidermis, parenchymatous cells of petioles or
secondary root phloem. Somatic embryos
arise from single cells located within the clusters of meristematic cells in
the callus or cell suspension. First
a pro-embryo is formed which then develops into an embryo, and finally a plant.
When
the somatic embryos develop directly on the excised plant (explant) without undergoing callus formation, it is referred to as direct somatic
embryogenesis (Fig 47.6A). This is
possible due to the presence of pre-embryonic determined cells (PEDQ found in
certain tissues of plants. The
characteristic features of direct somatic embryogenesis is avoiding the
possibility of introducing
somaclonal variations in the propagated plants.
In
indirect embryogenesis, the cells from explant (excised plant tissues) are made
to proliferate and form callus, from
which cell suspension cultures can be raised. Certain cells referred to as induced embryo genic determined cells (IEDC) from the cell
suspension can form somatic embryos.
Embryogenesis is made possible by the presence of growth regulators (in
appropriate concentration) and
under suitable environmental conditions.
Two
routes of somatic embryogenesis are known — direct and indirect
Indirect somatic embryogenesis is commercially very attractive since a large number of embryos can be generated in a small volume of culture medium. The somatic embryos so formed are synchronous and with good regeneration capability.
Artificial
seeds can be made by encapsulation of somatic embryos. The embryos, coated with sodium alginate and nutrient solution, are dipped in calcium
chloride solution. The calcium ions
induce rapid cross-linking of sodium alginate to produce small gel beads, each
containing an encapsulated embryo.
These artificial seeds (encapsulated embryos) can be maintained in a viable state till they are planted.
For a
successful in vitro clonal propagation (micro propagation), optimization of
several factors is needed.
Some of these factors are briefly described:
1.
Genotype of the plant:
Selection
of the right genotype of the plant species (by screening) is necessary for improved micro propagation. In general, plants with vigorous
germination and branching capacity are more
suitable for micro- propagation.
2. Physiological status of the explants:
Explants
(plant materials) from more recently produced parts of plants are more
effective than those from older
regions. Good knowledge of donor plants’ natural propagation process with special reference to growth stage and seasonal influence will
be useful in selecting explants.
3. Culture media:
The
standard plant tissue culture media are suitable for micro propagation during
stage I and stage II. However,
for stage III, certain modifications are required. Addition of growth regulators (auxins and cytokinins) and alterations in mineral
composition are required. This is
largely dependent on the type of culture (meristem, bud etc.).
4. Culture environment:
Light:
Photosynthetic
pigment in cultured tissues does absorb light and thus influence micro- propagation. The quality of light is also known to influence in
vitro growth of shoots, e.g blue
light induced bud formation in tobacco shoots. Variations in
diurnal illumination also influence
micro propagation. In general, an illumination of 16 hours day and
8 hours night is satisfactory for
shoot proliferation.
Temperature:
Majority of the culture for micro propagation requires an optimal temperature around 25°C. There are however, some exceptions e.g. Begonia X Cheimantha hybrid tissue grows at a low temperature (around 18°C).
Composition of gas phase:
The
constitution of the gas phase in the culture vessels also influences micro
propagation. Unorganized growth of
cells is generally promoted by ethylene, O2, CO2 ethanol and acetaldehyde.
Factors
Affecting in Vitro Rooting:
A
general description of the factors affecting micro propagation, particularly in
relation to shoot multiplication
is given above. For efficient in vitro rooting during micro- propagation, low concentration of salts (reduction to half to one quarter from
the original) is advantageous. Induction
of roots is also promoted by the presence of suitable auxin (NAA or IBA).
Micro
propagation has become a suitable alternative to conventional methods of vegetative propagation of plants. There are several advantages of
micro propagation.
Through
micro propagation, a large number of plants can be grown from a piece of plant tissue within a short period. Another advantage is that micro
propagation can be carried out throughout
the year, irrespective of the seasonal variations. Further, for many plants
that are highly resistant to
conventional propagation, micro propagation is the suitable alternative. The small sized propagules obtained in micro propagation can be easily
stored for many years (germplasm storage),
and transported across international boundaries.
It is
possible to produce disease-free plants through micro propagation. Meristem tip cultures are generally employed to develop pathogen-free plants
.In fact, micro propagation is successfully
used for the production of virus-free plants of sweet potato (Ipomea batatus), cassava (Manihot esculenta) and yam (Discorea rotundata).
Production of Seeds in Some Crops:
Micro
propagation, through axillary bud proliferation method, is suitable for seed production in some plants. This is required in certain plants
where the limitation for’ seed production
is high degree of genetic conservation e.g. cauliflower, onion.
Cost-effective Process:
Micro
propagation requires minimum growing space. Thus, millions of plant species can be maintained inside culture vials in a small room in a nursery.
The production cost is relatively
low particularly in developing countries (like India) where the
manpower and labour charges are low.
Automated Micro propagation:
It has now become possible to automate micro propagation at various stages. In fact, bio-reactors have been set up for large scale multiplication of shoots and bulbs. Some workers employ robots (in place of labourers) for micro- propagation, and this further reduces production cost of plants.
Disadvantages
of Micro propagation:
Contamination
of Cultures:
During
the course of micro propagation, several slow-growing microorganisms (e.g. Eswinia sp, Bacillus sp) contaminate and grow in cultures. The
microbial infection can be controlled
by addition of antibiotics or fungicides. However, this will adversely
influence propagation of plants.
Brewing of Medium:
Micro
propagation of certain plants (e.g. woody perennials) is often associated with accumulation of growth inhibitory substances in the medium.
Chemically, these substances are
phenolic compounds, which can turn the medium into dark colour.
Phenolic compounds are toxic and
can inhibit the growth of tissues. Brewing of the medium can be prevented by
the addition of ascorbic acid or citric
acid or polyvinyl pyrrolidone to the medium.
Genetic Variability:
When
micro propagation is carried out through shoot tip cultures, genetic
variability is very low. However, use
of adventitious shoots is often associated with pronounced genetic variability.
Vitrification:
During
the course of repeated in vitro shoot multiplication, the cultures exhibit
water soaked or almost
translucent leaves. Such shoots cannot grow and even may die. This phenomenon is referred to as vitrification. Vitrification may be
prevented by increasing the agar
concentration (from 0.6 to 1%) in the medium. However, increased
agar concentration reduces the
growth rate of tissues.
Cost Factor:
For
some micro propagation techniques, expensive equipment, sophisticated
facilities and trained manpower are
needed. This limits its use.
In vitro Micrografting
Micrografting
is an in vitro grafting technique which involves the placement of a meristem or shoot tip explant onto a decapitated rootstock that
has been grown aseptically from seed
or micropropagated cultures. Special techniques have been used for increasing
the percentage of
successful micrografts with the use of growth regulators, etiolation
treatments, antioxidants, higher
sucrose levels, silicon tubes, etc. The technique has great potential for improvement and large scale multiplication of fruit plants. It has
been used on commercial scale for
production of virus-free plants in fruit crops and viroid free plants.
Micrografting has also been used in prediction
of incompatibility between the grafting partners, histological studies, disease indexing, production of disease-free plants particularly
resistant to soil borne pathogens
and multiplication of difficult to root plants.
Stages
of micrografting
Micro-propagation
protocol for scion as well as rootstock needs to be standardized separately before performing the micrografting operation under in
vitroconditions. Thus, micrografting can be
divided into three main stages:
Establishment
and multiplication of scion
Shoot
or meristem tips intended for grafting can be taken from actively growing
shoots in greenhouse, chambers,
field or in vitro. Generally, apical shoot tips or nodal cuttings are used as explants for the establishment of in vitro cultures. Following
establishment, microshoots are transferred
to shoot proliferation medium where shoot number increases by the development
of new axillary shoots.
Microshoots of desired thickness, age and length are used as scions for in vitro grafting operations.
Establishment and multiplication of rootstock
Rootstocks
used for micrografting are in vitro or in vivo germinated seedlings and rooted or unrooted micropropagated shoots. When seedling rootstocks are
used and all stages of grafting are
conducted in vitro, seeds are surface sterilized and germinated aseptically in
vessels containing nutrient
salts. The seedlings may be supported on agar medium. Seedlings can also be on a porous substrate, such as sterile vermiculite, which allows
the growth of a branched root system.
Preparation of rootstock and scion for micrografting Micrografting is affected
by cutting off the top of the
seedling rootstocks usually just above the cotyledons or top of the micropropagated shoot and placing small shoot apices of scion onto
the exposed surface of decapitated rootstock
in such a way that the cambium layer or vascular ring of the cut surfaces coincides with each other. This is called surface placement
method. Wedge or cleft grafting is
performed, incase thickness of rootstock and scion material is
large enough to allow making of wedge
on the scion material. Firm contact between rootstock and scion is extremely
important at the graft junction for
proper union of partners and callus formation. Several techniques have been developed for holding grafts together until fusion takes
place such as translucent silicon
tubing, elastic strip, filter paper bridge, and glass tubing,
nylon bands, aluminum foil tubes, dual
layer apparatus of aluminum foil and absorbent paper. When grafts
are successful, rootstock and scion
grow together to produce a plant. It is usually necessary to examine freshly
grafted seedlings on a regular
basis and remove any adventitious shoot arising on or below the graft union.
Applications of micrografting
§ Virus
and viroid elimination
§ Production
of plants resistant to pests and diseases
§ Assessment
of graft incompatibility
§ Improvement
of plant regeneration
§ Mass
multiplication
§ Indexing
viral diseases
§ Safe
germplasm exchange
Production of Disease-Free Plants:
Many
plant species are infected with pathogens — viruses, bacteria, fungi,
mycoplasma and nematodes that cause systemic diseases. Although these diseases do
not always result in the death of plants, they
reduce the quality and yield of plants. The plants infected with bacteria and
fungi
frequently respond to chemical treatment by bactericides and fungicides. However, it is very difficult to cure the virus-infected plants.
Further, viral disease is easily transferred in
seed- propagated as well as vegetatively propagated plant species. Plant breeders are always interested to develop disease-free plants,
particularly viral disease-free plants.
This has become a reality through tissue cultures.
Apical Meristems with Low Concentration of Viruses:
In general, the apical meristems of the pathogen infected and disease harbouring plants are either free or carry a low concentration of viruses, for the following reasons:
- Absence of vascular tissue in the meristems through which viruses readily move in the plant body.
- Rapidly dividing meristematic cells with high metabolic activity do not allow viruses to multiply.
- Virus replication is inhibited by a high concentration of endogenous auxin in shoot apices.
Tissue culture techniques employing meristem-tips are successfully used for the production of disease-free plants, caused by several pathogens — viruses, bacteria, fungi, mycoplasmas.
Methods to Eliminate Viruses in Plants:
In
general, plants are infected with many viruses; the nature of some of them may
be unknown. The usage
virus-free plant implies that the given plant is free from all the viruses, although this may not be always true. The commonly used methods
for virus elimination in plants are listed
below, and briefly described next.
I.
Heat treatment of plant
II.
Meristem-tip culture
III.
Chemical treatment of media
IV.
Other in vitro methods
Heat
Treatment (Thermotherapy) of Plants:
In the
early days, before the advent of meristem cultures, in vivo eradication of
viruses from plants was
achieved by heat treatment of whole plants. The underlying principle is that many viruses in plant tissues are either partially or completely
inactivated at higher temperatures
with minimal injury to the host plant. Thermotherapy (at
temperatures 35-40°C) was carried out
by using hot water or hot air for elimination viruses from growing
shoots and buds.
1.
Most of the viruses are not sensitive to heat treatment.
2.
Many plant species do not survive after thermotherapy.
With
the above disadvantages, heat treatment has not become popular for virus
elimination.
Meristem-Tip Culture:
A
general description of the methodology adopted for meristem and shoot tip
cultures has been described. For
viral elimination, the size of the meristem used in cultures is very critical. This is due to the fact that most of the viruses exist by
establishing a gradient in plant tissues.
Meristem-tip cultures are influenced by the following factors:
- Physiological condition of the explant — actively growing buds are more effective.
- Thermotherapy prior to meristem-tip culture — for certain plants (possessing viruses in the meristematic regions), heat treatment is first given and then the meristem-tips are isolated and cultured.
- Culture medium —MS medium with low concentrations of auxins and cytokinins is ideal.
Chemical Treatment of Media:
Some
workers have attempted to eradicate viruses from infected plants by chemical treatment of the tissue culture media. The commonly used chemicals
are growth substances (e.g. cytokinins)
and antimetabolites (e.g thiouracil, acetyl salicylic acid).
There
are however, conflicting reports on the elimination viruses by chemical
treatment of the media. For
instance, addition of cytokinin suppressed the multiplication of certain
viruses while for some other
viruses, it actually stimulated.
Other in Vitro Methods:
Besides
meristem-tip culture, other in vitro methods are also used for raising
virus-free plants. In this regard
callus cultures have been successful to some extent. The callus derived from the infected tissue does not carry the pathogens throughout
the cells. In fact, the uneven distribution
of tobacco mosaic virus in tobacco leaves was exploited to develop virus-free
plants of tobacco. Somatic
cell hybridization, gene transformation and somaclonal variations also useful to raise disease-free plants.
Elimination of Pathogens Other than Viruses:
Besides
the elimination of viruses, meristem-tip cultures and callus cultures are also useful for eradication bacteria, fungi and mycoplasmas. Some
examples are given
1. The fungus Fusarium roseum has been successfully eliminated through meristem cultures from carnation plants.
2. Certain bacteria (Pseudomonas carophylli, Pectobacterium parthenii) are eradicated from carnation plants by using meristem cultures.
Merits and Demerits of Disease-Free Plant Production:
Among the culture techniques, meristem-tip culture is the most reliable method for virus and other pathogen elimination. This, however, requires good knowledge of plant pathology and tissue culture. Virus-free plants exhibit increased growth and vigour of plants, higher yield (e.g. potato), increased flower size (e.g. Chrysanthemum), and improved rooting of stem cuttings (e.g. Pelargonium) Virus-free plants are more susceptible to the same virus when exposed again. This is the major limitation. Reinfection of disease-free plants can be minimized with good knowledge of greenhouse maintenance.
Callus and Suspension Cultures
Callus
is an unorganized, proliferative mass of differentiated plant cells, and
usually occurs naturally as wound response. Tissues and cells cultured on an
agar-gelled medium form an unorganised mass of cells is also called callus. It
can be induced through culture of plant tissue on a medium usually containing
relatively high levels of auxin, especially 2,4-D.
However,
because of the phase of disorganization that occurs, plants regenerating from
callus, can be prone to genetic change.Callus cultures need to be sub-cultured
every 3-5 weeks in view of cell growth, nutrient depletion and medium drying.
Therefore, calluses are easy to maintain and are the most widely used. When
explants are cultured on a suitable PGR(s) combination, many of its cells
undergo division. Even mature and certain differentiated, e.g., parenchyma and
often colenchyma, cells undergo changes to become meristematic; this is called
dedifferentiation.
Dedifferentiation
involves, among other things, renewed and enhanced RNA and protein syntheses
leading to the formation of new cellular components needed for meristematic
activity. Initially, cell divisions are confined to the cut ends, but
subsequently it covers the entire explant. The resulting cell mass is
ordinarily unorganised, but it often consists of several cell types including
fibers, and vascular elements.
Suspension
Cultures:
Tissues
and cells cultured in a liquid medium produce a suspension of single cells and
cells clumps of few to many cells; these are called suspension cultures. Liquid
cultures must be constantly agitated, generally by a gyratory shaker at 100-250
rpm (revolution per minute), to facilitate aeration and dissociation of cell
clumps into smaller pieces.
Suspension
cultures grow much faster than callus cultures, need to be sub-cultured about
every week, allow a more accurate determination of the nutritional requirements
of cells and are the only system amenable to scaling up for a large-scale
production of cells and even somatic embryos (SEs). The suspension cultures are
broadly grouped as follows: (1) batch cultures, (2) continuous cultures, and
(3) immobilized cell cultures.
Batch
Cultures:
In
a batch culture, the same medium and all the cells produced are retained in the
culture vessel, e.g., culture flasks (100-250 ml), fermenters (variable size),
etc. The cell number or biomass of a batch culture exhibits a typical sigmoidal
curve, having a lag phase during which the cell number or biomass remains unchanged,
followed by a logarithmic (log) phase when there is a rapid increase in cell
number and, finally, ending in a stationary phase during which cell number does
not change.
The
lag phase duration depends mainly on inoculum size and growth phase of the culture
from which the inoculum is taken. The log phase lasts about 3-4 cell
generations (a cell generation is the time taken for doubling of cell number),
and the duration of a cell generation may vary from 22-48 hr, depending mainly
on the plant species. The stationary phase is forced on the culture by
depletion of the nutrients and possibly due to an accumulation of cellular
wastes. If the culture is kept in stationary phase for a prolonged period, the
cells may die.

Initiation of callus and suspension cultures
Batch
cultures are maintained by sub-culturing. They are used for initiation of cell
suspensions, which may be used for cloning, cell selection or as seed cultures
for scaling up or for continuous cultures.
They
are, however, unsuitable for studies on cell growth and metabolism because
there is a constant change in cell density and nutritional status of the
medium. But batch cultures are much more convenient than continuous cultures
and, as a result, are routinely used.
 A
Model curve for cell number in a batch culture
A
Model curve for cell number in a batch culture
Continuous
Cultures:
In
a continuous culture, the cell population is maintained in a steady state by
regularly replacing a portion of the used or spent medium by fresh medium. Such
culture systems are of either (1) closed or (2) open type. In a closed
continuous culture, cells are separated from the used medium taken out for
replacement, and added back to the culture so that cell biomass keeps on
increasing. In contrast, both cells and the used medium are taken out from open
continuous cultures and replaced by equal volume of fresh medium. The
replacement volume is so adjusted that cultures remain at submaximal growth
indefinitely.
The open cultures are of either turbidostat or chemostat types. In a turbidostat, cells are allowed to grow upto a preselected turbidity (usually, measured as OD) when a predetermined volume of the culture is replaced by fresh normal culture medium. But in a chemostat, a chosen nutrient is kept in a concentration so that it is depleted very rapidly to become growth limiting, while other nutrients are still in concentrations higher than required. In such a situation, any addition of the growth-limiting nutrient is reflected in cell growth. Chemostats are ideal for the determination of effects of individual nutrients on cell growth and metabolism.
Immobilized Cell Cultures:
Plant
cells and cell groups may be encapsulated in a suitable material, e.g., agarose
and calcium alginate gels, or entrapped in membranes or stainless steel
screens. The gel beads containing cells may be packed in a suitable column or,
alternatively, cells may be packed in a column of a membrane or wire cloth.
Liquid
medium is continuously run through the column to provide nutrients and aeration
to cells. Immobilization of cells changes their cellular physiology in
comparison to suspension culture cells; this offers several advantages for
their use in biochemical production, but they are usually not used for other
studies.
Subculture:
After
a period of time, it becomes necessary to transfer organs and tissues to fresh
media chiefly due to nutrient depletion and medium drying. This is particularly
true of tissue and cell cultures where a portion of tissue is used to inoculate
new culture tubes or flasks; this is known as sub-culturing. In general, callus
cultures are sub-cultured every 4-6 weeks, while suspension cultures need to be
sub-cultured every 3-14 days. Plant cell and tissue cultures may be maintained
indefinitely by serial sub-culturing.
In
case of suspension cultures, sub-culturing should be done about or somewhat
prior to the time of their maximum growth. The inoculums volume should be
20-25% of the fresh medium volume; in any case, the initial cell density of the
fresh culture (just after inoculation) should be around 5 x 104 cells m1-1 or
higher otherwise the cells may fail to divide.
Estimation
of Growth:
Cell
number is the most informative measure of cell growth. This measurement is
applicable to only suspension cultures, and even their cell aggregates must be
treated, e.g., with pectinase, to dissociate them into single cells before
counting the cell number in a haemocytometer.
Therefore,
cell number is estimated only where information obtained justifies the efforts.
In contrast, packed cell volume of suspension cultures is easily determined by
pipetting a known volume into a 15 ml graduated centrifuge tube, spinning at
200 ×g for 5 min and reading the volume of cell pellet, which is expressed as
ml cells/1 of culture.
Cells
from suspension cultures are filtered onto a filter paper or nylon filter,
washed with distilled water, excess water removed under vacuum and weighed
along with the filter; the filter is preweighed in wet condition. For dry
weight determination, the cells and the filter are dried in an oven at 60°C for
12 hr and weighed; the filter is pre-weighed in dry condition. Cell fresh and
dry weights may either be expressed as per ml (suspension culture) or per
culture.
Nuclear
Cytology:
Callus
and suspension cultures show both numerical (polyploidy and aneuploidy) and
structural (deletions, translocations, etc.) chromosome changes. The frequency
of these changes tends to increase with the duration of in vitro culture so
that some cultures may become predominantly or even completely polyploid or
aneuploid.
Explants
contain endopolyploid cells, which may give rise to a portion of the polyploid
cells in cultures. But most polyploid cells appear to originate through
endoreduplication (additional rounds of DNA replication without intervening
cell division) although selection for such cells cannot be ruled out.
Aneuploid
cells originate mainly due to anaphase irregularities like unequal chromatid
separation, lagging chromatids or chromosomes, anaphase bridges giving rise to
breakage-fusion-bridge cycle, chromosome fragmentation, etc.
The
cytogenetic status of cultured cells is influenced by several factors of the
culture system, e.g., GR concentrations and combination, culture age, liquid or
agar medium, subculture interval, sucrose concentration, etc. Suspension
cultures of many diploid species show a selection for diploid cells so that
they remain predominantly diploid for long periods, e.g., in case of Vicia
hajastana and Haplopappus gracilis cultures remained predominantly diploid for
over 300 days.
Secondary
metabolite production through Cell suspension cultures
Plant
cell and tissue cultures can be established routinely under sterile conditions
from explants, such as plant leaves, stems, roots, and meristems for
multiplication and extraction of secondary metabolites. Strain improvement,
methods for the selection of high-producing cell lines, and medium
optimizations can lead to an enhancement in secondary metabolite production.
The capacity for plant cell, tissue, and organ cultures to produce and accumulate many of the same valuable chemical compounds as the parent plant in nature has been recognized almost since the inception of in vitro technology. The strong and growing demand in today's marketplace for natural, renewable products has refocused attention on in vitro plant materials as potential factories for secondary phytochemical products and has paved the way for new research exploring secondary product expression in vitro. There is a series of distinct advantages to producing a valuable secondary product in plant cell culture, rather than in vivo in the whole crop plant.
These include the following:
- Production can be more reliable,
simpler, and more predictable.
- Isolation of the phytochemical can be rapid and efficient, when compared with extraction from complex whole plants
- Compounds produced in vitro
can directly parallel compounds in the whole plant.
- Interfering compounds that occur in
the field-grown plant can be avoided in cell cultures.
- Tissue and cell cultures can yield a source of defined standard phytochemicals in large volumes.
- Tissue and cell cultures are a
potential model to test elicitation.
- Cell cultures can be radiolabeled, such that the accumulated secondary products, when provided as feed to laboratory animals, can be traced metabolically.
While research to date has succeeded in producing a wide range of valuable secondary phytochemicals in unorganized callus or suspension cultures, in some cases production requires more differentiated micro plant or organ cultures. This situation often occurs when the metabolite of interest is only produced in specialized plant tissues or glands in the parent plant. A prime example is ginseng (Panax ginseng). Because saponin and other valuable metabolites are specifically produced in ginseng roots, root culture is required in vitro. Similarly, herbal plants such as Hypericum perforatum (St. John's wort), which accumulates the hypericins and hyperforins in foliar glands, have not demonstrated the ability to accumulate phytochemicals in undifferentiated cells. As another example, biosynthesis of lysine to anabasine occurs in tobacco (Nicotiana tabacum) roots, followed by the conversion of anabasine to nicotine in leaves. Callus and shoot cultures of tobacco can produce only trace amounts of nicotine because they lack the organ-specific compound anabasine. In other cases, at least some degree of differentiation in a cell culture must occur before a product can be synthesized (e.g., vincristine or vinblastine from Catharanthus roseus). Reliance of a plant on a specialized structure for production of a secondary metabolite, in some cases, is a mechanism for keeping a potentially toxic compound sequestered. Intensive activities have been centered on production of natural drugs or chemoprotective compounds from plant cell culture by one or more of the following strategies:
Accumulation
of secondary metabolites in plant cell cultures for plant cell culture
techniques to become economically viable, it is important to develop methods
that would allow for consistent generation of high yields of products from
cultured cells. Careful selection of productive cells and cultural conditions
resulted in accumulation of several products in higher levels in cultured
cells.
In order to obtain yields in high concentrations for commercial exploitation, efforts have focused on the stimulation of biosynthetic activities of cultured cells using various methods - Culture productivity is critical to the practical application of plant cell culture technology to production of plant-specific bioactive metabolites. Until now, various strategies have been developed to improve the production of secondary metabolites using plant cell cultures. The tissue culture cells typically accumulate large amounts of secondary compounds only under specific conditions. That means maximization of the production and accumulation of secondary metabolites by plant tissue cultured cells requires
- manipulating the parameters of the
environment and medium,
- selecting high yielding cell clones,
- precursor feeding, and
- elicitation.
Optimization
of cultural conditions:
Number
of chemical and physical factors like media components, phytohormones, pH,
temperature, aeration, agitation, light affecting production of secondary
metabolites has been extensively studied. Several products were found to be
accumulating in cultured cells at a higher level than those in native plants
through optimization of cultural conditions. Manipulation of physical aspects
and nutritional elements in a culture is perhaps the most fundamental approach
for optimization of culture productivity. For example, ginsenosides by Panax
ginseng, rosmarinic acid by Coleus bluemei, shikonin by Lithospermum
erythrorhizon, ubiquinone-10 by Nicotiana tabacum, berberin by Coptis japonica,
were accumulated in much higher levels in cultured cells than in the intact
plants.
Selection
of high-producing strains:
Plant
cell cultures represent a heterogeneous population in which physiological
characteristics of individual plant cells are different. Synthesis of several
products in high amounts using selection and screening of plant cell cultures
have been already described by many workers. Cell cloning methods provide a
promising way of selecting cell lines yielding increased levels of product.
Precursor
feeding:
Exogenous
supply of a biosynthetic precursor to culture medium may also increase the
yield of the desired product. This approach is useful when the precursors are
inexpensive. The concept is based on the idea that any compound, which is an
intermediate, in or at the beginning of a secondary metabolite biosynthetic
route, stands a good chance of increasing the yield of the final product.
Attempts to induce or increase the production of plant secondary metabolites,
by supplying precursor or intermediate compounds, have been effective in many
cases. For example, amino acids have been added to cell suspension culture
media for production of tropane alkaloids, indole alkaloids etc. Addition of
phenylalanine to Salvia officinalis cell suspension cultures stimulated the
production of rosmarinic acid. Addition of the same precursor resulted
stimulation of taxol production in Taxus cultures. Feeding ferulic acid to
cultures of Vanilla planifolia resulted in increase in vanillin accumulation.
Furthermore, addition of leucine, led to enhancement of volatile monoterpenes
in cultures of Perilla frutiscens, where as addition of geraniol to rose cell
cultures led to accumulation of nerol and citronellol.
Elicitation:
Plants
produce secondary metabolites in nature as a defense mechanism against attack
by pathogens. Elicitors are signals triggering the formation of secondary
metabolites. Use of elicitors of plant defense mechanisms, i.e. elicitation,
has been one of the most effective strategies for improving the productivity of
bioactive secondary metabolites. Biotic and abiotic elicitors which are
classified on their origin are used to stimulate secondary metabolite formation
in plant cell cultures, thereby reducing the process time to attain high
product concentrations. Production of many valuable secondary metabolites using
various elicitors were also reported
Conclusions
The
use of plant cell culture for the production of chemicals and pharmaceuticals
has made great strides building on advances in plant science. The increased use
of genetic tools and an emerging picture of the structure and regulation of
pathways for secondary metabolism will provide the basis for the production of
commercially acceptable levels of product. The increased appeal of natural
products for medicinal purposes coupled with the low product yields and supply
concerns of plant harvesation has renewed interest in large-scale plant cell
culture technology. Knowledge of biosynthetic pathways of desired compounds in
plants as well as in cultures is often still in its infancy, and consequently,
strategies are needed to develop an information based on a cellular and
molecular level. Because of the complex and incompletely understood nature of
plant cells in in vitro cultures, case-by-case studies have been used to
explain the problems occurring in the production of secondary metabolites from
cultured plant cells. These results show that plant cell culture systems have
potential for commercial exploitation of secondary metabolites. The
introduction of the techniques of molecular biology, so as to produce
transgenic cultures and to effect the expression and regulation of biosynthetic
pathways, is also likely to be a significant step towards making cell cultures
more generally applicable to the commercial production of secondary
metabolites.
Protoplast isolation, culture and regeneration
Protoplasts
are naked plant cells without the cell wall, but they possess plasma membrane
and all other cellular components. They represent the functional plant cells
but for the lack of the barrier, cell wall.
Protoplasts of different species
can be fused to generate a hybrid and this process is
referred to as
somatic hybridization (or protoplast
fusion). Cybridization is
the phenomenon of fusion of a normal protoplast with an enucleated
(without nucleus) protoplast that results in the formation of a cybrid or
cytoplast (cytoplasmic hybrids).
Historical
developments:
The
term protoplast was introduced in 1880 by Hanstein. The first isolation of
protoplasts was achieved by Klercker (1892) employing a mechanical method. A
real beginning in protoplast research was made in 1960 by Cocking who used an
enzymatic method for the removal of cell wall.
Rakabe
and his associates (1971) were successful to achieve the regeneration of whole
tobacco plant from protoplasts. Rapid progress occurred after 1980 in
protoplast fusion to improve plant genetic material, and the development of
transgenic plants.
Importance
of Protoplasts and Their Cultures:
The
isolation, culture and fusion of protoplasts is a fascinating field in plant
research. Protoplast isolation and their cultures provide millions of single
cells (comparable to microbial cells) for a variety of studies.
Protoplasts
have a wide range of applications; some of them are listed below:
- The protoplast in culture can be
regenerated into a whole plant.
- Hybrids can be developed from
protoplast fusion.
- It is easy to perform single cell
cloning with protoplasts.
- Genetic transformations can be
achieved through genetic engineering of protoplast DNA.
- Protoplasts are excellent materials
for ultra-structural studies.
- Isolation of cell organelles and
chromosomes is easy from protoplasts.
- Protoplasts are useful for membrane
studies (transport and uptake processes).
- Isolation of mutants from protoplast
cultures is easy.
Isolation
of Protoplasts:
Protoplasts
are isolated by two techniques
1) Mechanical
method
2) Enzymatic
method
Mechanical
Method:
Protoplast
isolation by mechanical method is a crude and tedious procedure. This results
in the isolation of a very small number of protoplasts.
The
technique involves the following stages:

- A small piece of epidermis from a
plant is selected.
- The cells are subjected to plasmolysis.
This causes protoplasts to shrink away from the cell walls.
- The tissue is dissected to release
the protoplasts.
Mechanical method for protoplast isolation is no more in use because of the following limitations:
- Yield of protoplasts and their viability
is low.
- It is restricted to certain tissues
with vacuolated cells.
- The method is laborious and tedious.
However,
some workers prefer mechanical methods if the cell wall degrading enzymes (of
enzymatic method) cause deleterious effects to protoplasts.
Enzymatic
Method:
Enzymatic
method is a very widely used technique for the isolation of protoplasts. The
advantages of enzymatic method include good yield of viable cells, and minimal
or no damage to the protoplasts.
Sources
of protoplasts:
Protoplasts
can be isolated from a wide variety of tissues and organs that include leaves,
roots, shoot apices, fruits, embryos and microspores. Among these, the
mesophyll tissue of fully expanded leaves of young plants or new shoots are
most frequently used. In addition, callus and suspension cultures also serve as
good sources for protoplast isolation.
Enzymes
for protoplast isolation:
Different
enzyme preparations are available in the market but the idea is to combine one
middle lamella dissolving and one cell wall digesting enzymes in proper
composition to achieve maximum protoplasts release from one gm. material.
Following
enzymes are used:
- Macerozymes R-10
- Cellulase – Onozuka R-10
- Hemicellulose
- Pectinase
- Drieselase
A
combination of these enzymes in a concentration of 0.5-2% is used. In many
cases only macerozyme and cellulase are sufficient to obtain protoplasts in
significant number. The enzyme solution (pH 5.5) is prepared in 10-15% sorbitol
or mannitol containing small amount of CaCl2 (7 mM) for membrane stability.
This
solution is sterilized through a membrane filter (cold sterilization) and leaf
or callus tissues are placed in it. Petri-plates containing tissue and enzyme
mixture are sealed with parafilm and incubated for 4-12 hours (sometimes 0.5 to
20 hrs.) on a rocking shaker at 24-26 °C.
After
incubation, solution is filtered through a wire or nylon mesh (50-100 µm) to
remove debris (undigested cells, tissues, broken cells etc.), transferred into
screw capped small centrifuge tubes (sterilized), and centrifuged at 100 g. The
protoplasts formed a pellet while the debris in the supernatant is carefully
removed.
Fresh
sterilized sorbitol solution (no enzyme) is added to tube and centrifuged. By
repeating the process 2 to 3 times, protoplasts are cleaned (debris is
removed). If 20% sucrose solution is used, protoplasts will float and debris
will settle during centrifugation at 200g for 1 min.
Floating
protoplasts are carefully removed with the help of sterilized pipette and
bulked together for further use. Protoplasts are counted by haemocytometer and
then diluted to proper strength (number per ml) in the culture medium
containing osmoticum.
The
enzymes that can digest the cell walls are required for protoplast isolation.
Chemically, the plant cell wall is mainly composed of cellulose, hemicellulose
and pectin which can be respectively degraded by the enzymes cellulose,
hemicellulose and pectinase. In fact, the various enzymes for protoplast
isolation are commercially available. The enzymes are usually used at a pH 4.5
to 6.0, temperature 25-30°C with a wide variation in incubation period that may
range from half an hour to 20 hours.
The
enzymatic isolation of protoplasts can be carried out by two approaches:
1.
Two step or sequential method:
The
tissue is first treated with pectinase (macerozyme) to separate cells by
degrading middle lamella. These free cells are then exposed to cellulose to
release protoplasts. Pectinase breaks up the cell aggregates into individual
cells while cellulose removes the cell wall proper.
2.
One step or simultaneous method:
This
is the preferred method for protoplast isolation. It involves the simultaneous
use of both the enzymes — macerozyme and cellulose.
Isolation
of protoplasts from leaves:
Leaves are most commonly used, for protoplast isolation, since it is possible to isolate uniform cells in large numbers.
The
procedure broadly involves the following steps:
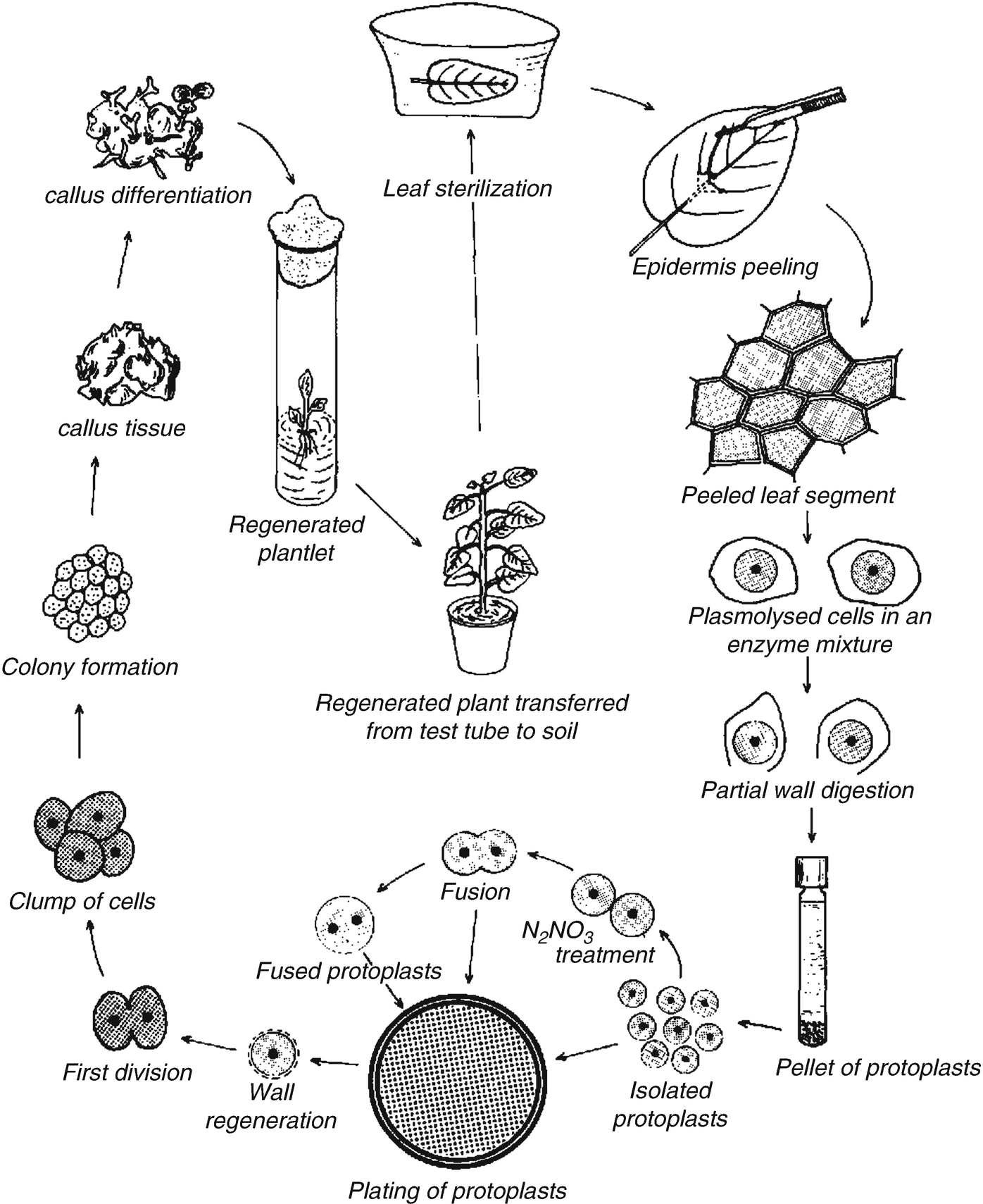
- Sterilization of leaves.
- Removal of epidermal cell layer.
- Treatment with enzymes.
- Isolation of protoplasts.
Besides
leaves, callus cultures and cell suspension cultures can also be used for the
isolation of protoplasts. For this purpose, young and actively growing cells
are preferred.
Purification
of protoplasts:
The
enzyme digested plant cells, besides protoplasts contain undigested cells,
broken protoplasts and undigested tissues. The cell clumps and undigested
tissues can be removed by filtration. This is followed by centrifugation and
washings of the protoplasts. After centrifugation, the protoplasts are
recovered above Percoll.
Viability
of protoplasts:
It
is essential to ensure that the isolated protoplasts are healthy and viable so
that they are capable of undergoing sustained cell divisions and regeneration.
There
are several methods to assess the protoplast viability:
- Fluorescein diacetate (FDA) staining
method—The dye accumulates inside viable protoplasts which can be detected
by fluorescence microscopy.
- Phenosafranine stain is selectively
taken up by dead protoplasts (turn red) while the viable cells remain
unstained.
- Exclusion of Evans blue dye by intact
membranes.
- Measurement of cell wall
formation—Calcofluor white (CFW) stain binds to the newly formed cell
walls which emit fluorescence.
- Oxygen uptake by protoplasts can be
measured by oxygen electrode.
- Photosynthetic activity of
protoplasts.
- The ability of protoplasts to undergo
continuous mitotic divisions (this is a direct measure).
Culture
of Protoplasts:
The
very first step in protoplast culture is the development of a cell wall around
the membrane of the protoplast. This is followed by the cell divisions that
give rise to a small colony. With suitable manipulations of nutritional and
physiological conditions, the cell colonies may be grown continuously as
cultures or regenerated to whole plants. Protoplasts are cultured either in
semisolid agar or liquid medium. Sometimes, protoplasts are first allowed to
develop cell wall in liquid medium, and then transferred to agar medium.
Agar
culture:
Agarose
is the most frequently used agar to solidify the culture media. The
concentration of the agar should be such that it forms a soft agar gel when
mixed with the protoplast suspension. The plating of protoplasts is carried out
by Bergmann’s cell plating technique .In agar cultures, the protoplasts remain
in a fixed position, divide and form cell clones. The advantage with agar
culture is that clumping of protoplasts is avoided.
Liquid
culture:
Liquid
culture is the preferred method for protoplast cultivation for the following
reasons:
- It is easy to dilute and transfer.
- Density of the cells can be
manipulated as desired.
- For some plant species, the cells
cannot divide in agar medium, therefore liquid medium is the only choice.
- Osmotic pressure of liquid medium can
be altered as desired.
Culture Media:
The
culture media with regard to nutritional components and osmoticum are briefly
described.
Nutritional
components:
In
general, the nutritional requirements of protoplasts are similar to those of
cultured plant cells (callus and suspension cultures). Mostly, MS and B5 media
with suitable modifications are used.
Some of the special features of protoplast culture media are listed below:
- The medium should be devoid of
ammonium, and the quantities of iron and zinc should be less.
- The concentration of calcium should
be 2-4-times higher than used for cell cultures. This is needed for
membrane stability.
- High auxin/kinetin ratio is suitable
to induce cell divisions while high kinetin/auxin ratio is required for
regeneration.
- Glucose is the preferred carbon source
by protoplasts although a combination of sugars (glucose and sucrose) can
be used.
- The vitamins used for protoplast
cultures are the same as used in standard tissue culture media.
Osmoticum
and osmotic pressure:
Osmoticum broadly refers to the reagents/ chemicals that are added to increase the osmotic pressure of a liquid. The isolation and culture of protoplasts require osmotic protection until they develop a strong cell wall. In fact, if the freshly isolated protoplasts are directly added to the normal culture medium, they will burst. Thus, addition of an osmoticum is essential for both isolation and culture media of protoplast to prevent their rupture. The osmotica are of two types — non-ionic and ionic.
Non-ionic
osmotica:
The
non-ionic substances most commonly used are soluble carbohydrates such as
mannitol, sorbitol, glucose, fructose, galactose and sucrose. Mannitol, being
metabolically inert, is most frequently used.
Ionic
osmotica:
Potassium
chloride, calcium chloride and magnesium phosphate are the ionic substances in
use to maintain osmotic pressure. When the protoplasts are transferred to a
culture medium, the use of metabolically active osmotic stabilizers (e.g.,
glucose, sucrose) along with metabolically inert osmotic stabilizers (mannitol)
is advantageous. As the growth of protoplasts and cell wall regeneration
occurs, the metabolically active compounds are utilized, and this results in
the reduced osmotic pressure so that proper osmolarity is maintained.
Culture
Methods:
The
culture techniques of protoplasts are almost the same that are used for cell
culture with suitable modifications. Some important aspects are briefly given.
Feeder
layer technique:
For
culture of protoplasts at low density feeder layer technique is preferred. This
method is also important for selection of specific mutant or hybrid cells on
plates. The technique consists of exposing protoplast cell suspensions to
X-rays (to inhibit cell division with good metabolic activity) and then plating
them on agar plates.
Co-culture
of protoplasts:
Protoplasts
of two different plant species (one slow growing and another fast growing) can
be co- cultured. This type of culture is advantageous since the growing species
provide the growth factors and other chemicals which help in the generation of
cell wall and cell division.
The
co-culture method is generally used if the two types of protoplasts are
morphologically distinct.
Micro
drop culture:
Specially
designed dishes namely cuprak dishes with outer and inner chambers are used for
micro drop culture. The inner chamber carries several wells wherein the
individual protoplasts in droplets of nutrient medium can be added. The outer
chamber is filled with water to maintain humidity. This method allows the
culture of fewer protoplasts for droplet of the medium.
Regeneration
of Protoplasts:
Protoplast regeneration which may also be regarded as protoplast development occurs in two stages:
- Formation of cell wall.
- Development of callus/whole plant.
Formation
of cell wall:
The
process of cell wall formation in cultured protoplasts starts within a few
hours after isolation that may take two to several days under suitable
conditions. As the cell wall development occurs, the protoplasts lose their
characteristic spherical shape. The newly developed cell wall by protoplasts
can be identified by using calcofluor white fluorescent stain.
The freshly formed cell wall is composed of loosely bound micro fibrils which get organized to form a typical cell wall. This process of cell wall development requires continuous supply of nutrients, particularly a readily metabolised carbon source (e.g. sucrose). Cell wall development is found to be improper in the presence of ionic osmotic stabilizers in the medium. The protoplasts with proper cell wall development undergo normal cell division. On the other hand, protoplasts with poorly regenerated cell wall show budding and fail to undergo normal mitosis.
Development
of Callus/whole Plant:
As
the cell wall formation around protoplasts is complete, the cells increase in
size, and the first division generally occurs within 2-7 days. Subsequent
divisions result in small colonies, and by the end of third week, visible
colonies (macroscopic colonies) are formed. These colonies are then transferred
to an osmotic-free (mannitol or sorbitol-free) medium for further development
to form callus. With induction and appropriate manipulations, the callus can
undergo organogenic or embryo genic differentiation to finally form the whole
plant. A general view of the protoplast isolation, culture and regeneration is
represented in Fig given below.
Plant
regeneration can be done from the callus obtained either from protoplasts or
from the culture of plant organs. There are however, certain differences in
these two calluses. The callus derived from plant organs carries preformed buds
or organized structures, while the callus from protoplast culture does not have
such structures.
Sub-Protoplasts:
The
fragments derived from protoplasts that do not contain all the contents of
plant cells are referred to as sub-protoplasts. It is possible to
experimentally induce fragmentation of protoplasts to form sub-protoplasts.
This can be done by application of different centrifugal forces created by
discontinuous gradients during centrifugation. Exposure of protoplasts to
cytochalasin B in association with centrifugation is a better approach for
fragmentation of protoplasts.
There
are three types of sub-protoplasts:

1.
Mini-protoplasts:
These
are also called as karyoplasts and contain the nucleus. Mini-protoplasts can
divide and are capable of regeneration into plants.
2.
Cytoplasts:
These
are sub-protoplasts containing the original cytoplasmic material (in part or
full) but lack nucleus. Thus, cytoplasts are nuclear-free sub-protoplasts which
cannot divide, but they can be used for cybridization.
3.
Micro-protoplasts:
This
term was suggested for sub-protoplasts that contain not all but a few
chromosomes.
Somatic
Hybridization: Aspects, Applications and Limitations
The
conventional method to improve the characteristics of cultivated plants, for
years, has been sexual hybridization. The major limitation of sexual
hybridization is that it can be performed within a plant species or very
closely related species. This restricts the improvements that can be done in
plants.
The
species barriers for plant improvement encountered in sexual hybridization can
be overcome by somatic cell fusion that can form viable hybrids. Somatic
hybridization broadly involves in vitro fusion of isolated protoplasts to form
a hybrid cell and its subsequent development to form a hybrid plant.
Plant protoplasts are of immense utility in somatic plant cell genetic manipulations and improvement of crops. Thus, protoplasts provide a novel opportunity to create cells with new genetic constitution. And protoplast fusion is a wonderful approach to overcome sexual incompatibility between different species of plants. More details on the applications of somatic hybridization are given later.
Somatic hubridization involves the following aspects:
- Fusion of protoplasts
- Selection of hybrid cells
- Identification of hybrid plants.
A.
Fusion of Protoplasts:
As
the isolated protoplasts are devoid of cell walls, there in vitro fusion
becomes relatively easy. There are no barriers of incompatibility (at
interspecific, inter-generic or even at inter-kingdom levels) for the
protoplast fusion. Protoplast fusion that involves mixing of protoplasts of two
different genomes can be achieved by spontaneous, mechanical, or induced fusion
methods.
Spontaneous
fusion:
Cell
fusion is a natural process as is observed in case of egg fertilization. During
the course of enzymatic degradation of cell walls, some of the adjoining
protoplasts may fuse to form homokaryocytes (homokaryons). These fused cells
may sometimes contain high number of nuclei (2-40).
This
is mainly because of expansion and subsequent coalescence of plasmodermal
connections between cells. The frequency of homokaryon formation was found to
be high in protoplasts isolated from dividing cultured cells. Spontaneously
fused protoplasts, however, cannot regenerate into whole plants, except
undergoing a few cell divisions.
Mechanical
fusion:
The
protoplasts can be pushed together mechanically to fuse. Protoplasts of Lilium
and Trillium in enzyme solutions can be fused by gentle trapping in a
depression slide. Mechanical fusion may damage protoplasts by causing injuries.
Induced
fusion:
Freshly
isolated protoplasts can be fused by induction. There are several
fusion-inducing agents which are collectively referred to as fusogens e.g.
NaN03, high pH/Ca2+, polyethylene glycol, polyvinyl alcohol, lysozyme,
concavalin A, dextran, dextran sulfate, fatty acids and esters, electro fusion.
Some of the fusogens and their use in induced fusion are described.
A
diagrammatic representation of protoplast fusion is given below
The
isolated protoplasts are exposed to a mixture of 5.5% NaNO3 in 10% sucrose
solution. Incubation is carried out for 5 minutes at 35°C, followed by
centrifugation (200 x g for 5 min). The protoplast pellet is kept in a water
bath at 30°C for about 30 minutes, during which period protoplast fusion
occurs. NaNO3 treatment results in a low frequency of heterokaryon formation,
particularly when mesophyll protoplasts are fused.
High
pH and high Ca2+ ion treatment:
This
method was first used for the fusion of tobacco protoplasts, and is now in use
for other plants also. The method consists of incubating protoplasts in a
solution of 0.4 M mannitol containing 0.05 M CaCI2 at pH 10.5 (glycine-NaOH
buffer) and temperature 3 7°C for 30-40 minutes. The protoplasts form
aggregates, and fusion usually occurs within 10 minutes. By this method, 20-50%
of the protoplasts are involved in fusion.
Polyethylene
glycol (PEG) treatment:
This
has become the method of choice, due to its high success rate, for the fusion
of protoplasts from many plant species. The isolated protoplasts in culture
medium (1 ml) are mixed with equal volume (1 ml) of 28-56% PEG (mol. wt. 1500-6000
Daltons) in a tube. PEG enhances fusion of protoplasts in several species. This
tube is shaken and then allowed to settle.
The
settled protoplasts are washed several times with culture medium.
PEG
treatment method is widely used protoplast fusion as it has several advantages:
- It results in a reproducible
high-frequency of heterokaryon formation.
- Low toxicity to cells.
- Reduced formation of bi-nucleate
heterokaryons.
- PEG-induced fusion is non-specific
and therefore can be used for a wide range of plants.
Electro-fusion:
In
this method, electrical field is used for protoplast fusion. When the
protoplasts are placed in a culture vessel fitted with micro- electrodes and an
electrical shock is applied, protoplasts are induced to fuse. Electro-fusion
technique is simple, quick and efficient and hence preferred by many workers.
Further,
the cells formed due to electro-fusion do not show cytotoxic responses as is
the case with the use of fusogens (including PEG). The major limitation of this
method is the requirement of specialized and costly equipment.
Mechanism
of fusion:
The
fusion of protoplasts involves three phases agglutination, plasma membrane
fusion and formation of heterokaryons.
1.
Agglutination (adhesion):
When
two protoplasts are in close contact with each other, adhesion occurs.
Agglutination can be induced by fusogens e.g. PEG, high pH and high Ca2+.
2.
Plasma membrane fusion:
Protoplast
membranes get fused at localized sites at the points of adhesion. This leads to
the formation of cytoplasmic bridges between protoplasts. The plasma membrane
fusion can be increased by high pH and high Ca2+, high temperature and PEC, as
explained below.
a) High
pH and high Ca2+ ions neutralize the surface charges on the protoplasts. This
allows closer contact and membrane fusion between agglutinated protoplasts.
b) High
temperature helps in the intermingling of lipid molecules of agglutinated
protoplast membranes so that membrane fusion occurs.
c) PEG
causes rapid agglutination and formation of clumps of protoplasts. This results
in the formation of tight adhesions of membranes and consequently their fusion.
3.
Formation of heterokaryons:
The
fused protoplasts get rounded as a result of cytoplasmic bridges leading to the
formation of spherical homokaryon or heterokaryon.
B.
Selection of Hybrid Cells:
About
20-25% of the protoplasts are actually involved in the fusion. After the fusion
process, the protoplast population consists of a heterogenous mixture of
un-fused chloroplasts, homokaryons and heterokaryons as shown below. It is
therefore necessary to select the hybrid cells (heterokaryons). The commonly
used methods employed for the selection of hybrid cells are biochemical, visual
and cytometric methods.
Biochemical
methods:
The
biochemical methods for selection of hybrid cells are based on the use of
biochemical compounds in the medium (selection medium). These compounds help to
sort out the hybrid and parental cells based on their differences in the
expression of characters.
Drug sensitivity and auxotrophic mutant selection methods are described below:
1.
Drug sensitivity:
This
method is useful for the selection hybrids of two plant species, if one of them
is sensitive to a drug. Protoplasts of Petunia hybride (species A) can form
macroscopic callus on MS medium, but are sensitive to (inhibited by)
actinomycin D. Petunia parodii protoplasts (species B) form small colonies, but
are resistant to actinomycin D.
When
these two species are fused, the fused protoplasts derive both the characters —
formation of macroscopic colonies and resistance to actinomycin D on MS medium.
This helps in the selection of hybrids. The parental protoplasts of both the
species fail to grow. Protoplasts of P. parodii form very small colonies while
that of P. hybrida are inhibited by actinomycin D.

2.
Auxotrophic mutants:
Auxotroph’s
are mutants that cannot grow on a minimal medium and therefore require specific
compounds to be added to the medium. Nitrate reductase deficient mutants of
tobacco (N. tabacum) are known. The parental protoplasts of such species cannot
grow with nitrate as the sole source of nitrogen while the hybrids can grow.
Two
species of nitrate reductase deficiency— one due to lack of apoenzyme (nia-type
mutant) and the other due to lack of molybdenum cofactor (cnx- type mutant) are
known. The parental protoplasts cannot grow on nitrate medium while the hybrid
protoplasts can grow (Fig.44.7).

The selection of auxotrophic mutants is possible only if the hybrid cells can grow on a minimal medium. Another limitation of the technique is the paucity of higher plant auxotroph’s.
Visual
methods:
Visual
selection of hybrid cells, although tedious is very efficient. In some of the
somatic hybridization experiments, chloroplast deficient (albino or non-green)
protoplasts of one parent are fused with green protoplasts of another parent.
This
facilitates the visual identification of haterokaryons under light microscope.
The heterokaryons are bigger and green in colour while the parental protoplasts
are either small or colourless. Further identification of these heterokaryons
has to be carried out to develop the specific hybrid plant. There are two
approaches in this direction — growth on selection medium, and mechanical
isolation.
1.
Visual selection coupled with differential media growth:
There
exist certain natural differences in the sensitivity of protoplasts to the
nutrients of a given medium. Thus, some media can selectively support the
development of hybrids but not the parental protoplasts. A diagrammatic
representation of visual selection coupled with the growth of heterokaryons on
a selection medium is given below.

The
visually identified heterokaryons under the microscope can be isolated by
mechanical means. This involves the use of a special pipette namely Drummond
pipette. The so isolated heterokaryons can be cloned to finally produce somatic
hybrid plants. The major limitation of this method is that each type of hybrid
cell requires a special culture medium for its growth. This can be overcome by
employing micro drop culture of single cells using feeder layers.
Cytometric
methods:
Some
workers use flow cytometry and fluorescent-activated cell sorting techniques
for the analysis of plant protoplasts while their viability is maintained. The
same techniques can also be applied for sorting and selection of heterokaryons.
The hybrid cells derived from such selections have proved useful for the
development of certain somatic hybrid plants.
C.
Identification of Hybrid (Cells) Plants:
The
development of hybrid cells followed by the generation of hybrid plants
requires a clear proof of genetic contribution from both the parental
protoplasts. The hybridity must be established only from euploid and not from
aneuploid hybrids. Some of the commonly used approaches for the identification
of hybrid plants are briefly described.
Morphology
of hybrid plants:
Morphological
features of hybrid plants which usually are intermediate between two parents
can be identified. For this purpose, the vegetative and floral characters are
considered. These include leaf shape, leaf area, root morphology, flower shape,
its structure, size and colour, and seed capsule morphology.
The somatic hybrids such as pomatoes and topatoes which are the fused products of potato and tomato show abnormal morphology, and thus can be identified. Although the genetic basis of the morphological characters has not been clearly known, intermediate morphological features suggest that the traits are under the control of multiple genes. It is preferable to support hybrid morphological characters with evidence of genetic data.
Isoenzyme
analysis of hybrid plants:
The
multiple forms of an enzyme catalysing the same reaction are referred to as
isoenzymes. Electrophoretic patterns of isoenzymes have been widely used to
verify hybridity. Somatic hybrids possess specific isoenzymes (of certain
enzymes) of one or the other parent or both the parents simultaneously.
There
are many enzymes possessing unique isoenzymes that can be used for the
identification of somatic hybrids e.g. amylase, esterase, aspartate
aminotransferase, phosphodiesterase, isoperoxidase, and hydrogenases (of
alcohol, lactate, malate). If the enzyme is dimeric (having two subunits),
somatic hybrids usually contain an isoenzyme with an intermediate mobility
properties. The isoenzymes are often variable within the same plant. Therefore,
it is necessary to use the same enzyme from each plant (parents and somatic
hybrids), from a specific tissue with the same age.
Chromosomal
constitution:
The
number of chromosomes present in the hybrid cells can be directly counted. This
provides information on the ploidy state of the cells. The somatic hybrids are
expected to possess chromosomes that are equal to the total number of chromosomes
originally present in the parental protoplasts. Sometimes, the hybrids are
found to contain more chromosomes than the total of both the parents. The
presence of chromosomal markers is greatly useful for the genetic analysis of
hybrid cells.
Molecular
techniques:
Many
recent developments in molecular biology have improved the understanding of
genetic constitution of somatic plant hybrids.
Some
of them are listed below:
- Differences in the restriction
patterns of chloroplast and mitochondrial DNAs.
- Molecular markers such as RFLP, AFLP,
RAPD and microsatellites.
- PCR technology.
Chromosome
Number in Somatic Hybrids:
The
chromosome number in the somatic hybrids is generally more than the total
number of both of the parental protoplasts.
However,
wide variations are reported which may be due to the following reasons:
- Fusion of more than two protoplasts.
- Irregularities in mitotic cell divisions.
- In fusogen or electro-induced fusions, about one third of the fusions occur between morethan two protoplasts.
- Differences in the status of protoplasts (actively dividing or quiescent) from the two species of plants result in formation of asymmetric hybrids.
- Asymmetric hybrids may be due to unequal replication of DNA in the fusing protoplasts.
- Protoplast isolation and culture may also lead to somaclonal variations, and thus variations in chromosome number.
A
selected list of interspecific hybrids produced through protoplast fusion along
with the number of chromosomes in the hybrids is given below

Symmetric and asymmetric hybrids:
If
the chromosome number in the hybrid is the sum of the chromosomes of the two
parental protoplasts, the hybrid is said to be symmetric. Symmetric hybrids
between incompatible species are usually sterile. This may be due to production
of 3n hybrids by fusing 2n of one species with n of another species.
Asymmetric
hybrids have abnormal or wide variations in the chromosome number than the
exact total of two species. These hybrids are usually formatted with full
somatic complement of one parental species while all or nearly all of the
chromosomes of other parental species are lost during mitotic divisions.
Asymmetric hybrids may be regarded as cybrids but for the introgressed genes.
Cybrids:
The
cytoplasmic hybrids where the nucleus is derived from only one parent and the
cytoplasm is derived from both the parents are referred to as cybrids. The
phenomenon of formation of cybrids is regarded as cybridization. Normally,
cybrids are produced when protoplasts from two phytogenetically distinct species
are fused. Genetically, cybrids are hybrids only for cytoplasmic traits.
Hybrids
and Somatic Incompatibility:
Many
a times, production of full-pledged hybrids through fusion of protoplasts of
distantly related higher plant species is rather difficult due to instability
of the two dissimilar genomes in a common cytoplasm. This phenomenon is
referred to as somatic incompatibility. Hybrids formed despite somatic
incompatibility may exhibit structural and developmental abnormalities. Several
generations may be required to eliminate the undesirable genes. Due to this
limitation in somatic hybridization, cybridization involving protoplast fusion
for partial genome transfer is gaining importance in recent years.
Methodology
of Cybridization:
A
diagrammatic representation of the formation of hybrids and cybrids is given in
below

As the formation of heterokaryon occurs during hybridization, the nuclei can be stimulated to segregate so that one protoplast contributes to the cytoplasm while the other contributes nucleus alone (or both nucleus and cytoplasm). In this way cybridization can be achieved.
Some
of the approaches of cybridization are given here under:
- The protoplasts of cytoplasm donor
species are irradiated with X-rays or ? -rays. This treatment renders the
protoplasts inactive and non-dividing, but they are efficient donors of
cytoplasmic constituents when fused with recipient protoplasts.
- Normal protoplasts can be directly
fused with enucleated protoplasts. Enucleated protoplasts can be isolated
by high-speed centrifugation.
- Protoplasts are inactivated by
metabolic inhibitors such as iodoacetate. In practice, iodoacetate treated
protoplasts are fused with X-rays irradiated protoplasts for more
efficient formation of cybrids.
- It is possible to suppress nuclear
division in some protoplasts and fuse them with normal protoplasts.
Genetic
recombination in Asexual or Sterile Plants:
There
are many plants that cannot reproduce sexually. Somatic hybridization is a
novel approach through which two parental genomes of a sexual or sterile plants
can be brought together. Thus, by fusing parental protoplasts, fertile diploids
and polyploidy can be produced.
Overcoming
Barriers of Sexual Incompatibility:
Sexual
crossing between two different species (interspecific) and two different genus
(inter-generic) is impossible by conventional breeding methods. Somatic
hybridization overcomes the sexual incompatibility barriers.
Two
examples are given hereunder:
- Fusion between protoplasts of potato
(Solanum tuberosum) and tomato (Lycopersicon esculentum) has created
pomato (Solanopersicon, a new genus).
- Interspecific fusion of four different species of rice (Oryza brachyantha, O. elchngeri, O. officinalis and O. perrieri) could be done to improve the crop.
A
list of selected examples of somatic hybrids developed by interspecific
protoplast fusion is given in the following table

A
Novel Approach for Gene Transfer:
Somatic
hybridization has made it possible to transfer several desirable genetic
characters among the plants
Applications
of Cybrids:
Cybridization is a wonderful technique wherein the desired cytoplasm can be transferred in a single step. Cybrids are important for the transfer of cytoplasmic male sterility (CMS), antibiotic and herbicide resistance in agriculturally useful plants. Some of the genetic traits in certain plants are cytoplasmically controlled. This includes some types of male sterility, resistance to certain antibiotics and herbicides.
Cybridization
has been successfully used to transfer CMS in rice. Cybrids of Brassica
raphanus that contain nucleus of B. napus, chloroplasts of atrazinc
resistant B. campestris and male sterility from Raphanus sativas
have been developed.
Applications
of Somatic Hybridization:
Somatic
hybridization has opened new possibilities for the in vitro genetic
manipulation of plants to improve the crops.
Some
of the practical applications are briefly given:
1.
Disease resistance:
Several
interspecific and inter-generic hybrids with disease resistance have been created.
Many disease resistance genes (e.g., tobacco mosaic virus, potato virus X, club
rot disease) could be successfully transferred from one species to another. For
example, resistance has been introduced in tomato against diseases such as TMV,
spotted wilt virus and insect pests.
2.
Environmental tolerance:
The
genes responsible for the tolerance of cold, frost and salt could be
successfully introduced through somatic hybridization, e.g., introduction of
cold tolerance gene in tomato.
3.
Quality characters:
Somatic
hybrids for the production of high nicotine content, and low erucic acid have
been developed.
A selected list of genetic traits transferred through protoplast fusion in crop plant species

4.
Cytoplasmic male sterility:
A
modification of hybridization in the form of cybridization has made it possible
to transfer cytoplasmic male sterility.
Other
Application of Somatic Hybridization:
1.
Somatic hybridization has helped to study the cytoplasmic genes and their
functions. In fact, the information is successfully used in plant breeding
programmes.
2.
Protoplast fusion will help in the combination of mitochondria and chloroplasts
to result in a unique nuclear-cytoplasmic genetic combination.
3.
Somatic hybridization can be done in plants that are still in juvenile phase.
4.
Protoplast transformation (with traits like nitrogen fixation by incorporating
exogenous DNA) followed by somatic hybridization will yield innovative plants.
Limitations
of Somatic Hybridization:
Although
somatic hybridization is a novel approach in plant biotechnology, there are
several problems and limitations.
The
success of the technique largely depends on overcoming these limitations, some
of
which
are listed below:
- Somatic, hybridization does not
always produce plants that give fertile and visible seeds.
- Regenerated plants obtained from
somatic hybridization are often variable due to somaclonal variations,
chromosomal elimination, organelle segregation etc.
- Protoplast culture is frequently
associated with genetic instability.
- Protoplast fusion between different
species/genus is easy, but the production of viable somatic hybrids is not
possible in all instances.
- Some of the somatic hybrids, particularly
when produced by the fusion of taxonomically different partners, are
unbalanced and not viable.
- There are limitations in the
selection methods of hybrids, as many of them are not efficient.
- There is no certainty as regards the
expression of any specific character in somatic hybridization.
- Somatic hybridization between two
diploids results in the formation of an amphidiploid which is not
favourable. For this reason, haploid protoplasts are recommended in
somatic hybridization.
Comments
Post a Comment